
1-broMo-4-phthaldehyde synthesis
- Product Name:1-broMo-4-phthaldehyde
- CAS Number:50672-84-9
- Molecular formula:C11H7BrO
- Molecular Weight:235.08
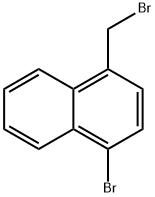
79996-99-9
50672-84-9
318 g of 1-bromo-4-bromomethylnaphthalene (II), 371.5 g of hexamethylenetetetramine, 795 g of glacial acetic acid, and 795 g of water were added to a 3L three-necked flask and reacted at reflux for 3 h at 100 °C. The reaction process was monitored by thin layer chromatography (TLC). Upon completion of the reaction, 636 g of concentrated hydrochloric acid was rapidly added dropwise for 0.5 hr. After the reaction mixture was cooled to room temperature, 4 L of water was added and stirred continuously for 3 hours. The solid product was collected by filtration, pulped with 2 L of water for 2 hours, filtered again, and the filter cake was washed twice with water, drained, and dried at 40 °C. The dried product was recrystallized with 318 g of ethanol and finally dried to give 200.4 g of 4-bromo-1-naphthaldehyde (III) in 80.4% yield. The molar ratio of 1-bromo-4-bromomethylnaphthalene (II) to hexamethylenetetetramine in the reaction was 1:2.5.
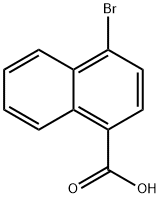
16650-55-8
153 suppliers
$12.00/1g
50672-84-9
98 suppliers
$7.00/100mg
Yield: 78%
Reaction Conditions:
Stage #1:4-bromonaphthalene-1-carboxylic acid with diborane in tetrahydrofuran at 0 - 25; for 5 h;Inert atmosphere;
Stage #2: with sodium hydrogencarbonate;Dess-Martin periodane in tetrahydrofuran;dichloromethane at 0; for 4 h;Inert atmosphere;
Steps:
3.2.1 4-Bromo-1-naphthaldehyde (7)
BH3 (40.0 mL, 1 mol L-1) was added to a solution of 7a (5.0 g, 20.0 mmol) in dry THF (50.0 mL) at 0°C, and the mixture stirred at room temperature for 5 h. The reaction was quenched with a saturated aqueous solutionof NH4Cl and concentrated under vacuum. The aqueous residue was extracted with EtOAc (3 × 200.0 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The residue was used in the next step without purification. Dess-Martin reagent (8.5 g, 20.0 mmol) and NaHCO3 (5.0 g, 60.0 mmol) were added to a solution of the residue in CH2Cl2 (150.0 mL) at 0°C. The reaction mixture was stirred at 0°C for 4 h, H2O(200.0 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 200.0 mL). The combined organic layer was concentrated under vacuum. The residue was purified by chromatography (EtOAc/hexanes = 1:20) to afford the compound 7 (yield 3.6 g, 78%) as a colorless solid; m.p. 79-81°C. 1H NMR (400 MHz, CDCl3): δ = 10.36 (s, 1H,-CHO), 9.27 (d, J = 8.4 Hz, 1H, Ar-H), 8.35 (d, J = 8.4 Hz, 1H,Ar-H), 7.95 (d, J = 7.6 Hz, 1H, Ar-H), 7.79 (d, J = 7.6 Hz, 1H,Ar-H), 7.69-7.75 (m, 2H, Ar-H). 13C NMR (100 MHz, CDCl3):δ = 192.6, 136.1, 132.1, 131.4, 131.3, 130.9, 129.8, 129.3, 128.3,127.7, 125.1. HRMS ((+)-ESI): m/z = 234.9731. (calcd. 234.9753 for C11H7BrO, [M + H]+).
References:
Li, Xiaowan;Si, Tongxu;Ku, Chuenfai;Zhang, Hongjie;Wang, Mingzhong;Chan, Albert S.C. [Zeitschrift fur Naturforschung, B: Chemical Sciences,2019,vol. 74,# 2,p. 183 - 189]
79996-99-9
51 suppliers
inquiry
50672-84-9
98 suppliers
$7.00/100mg

56052-26-7
40 suppliers
$60.00/25 mg
50672-84-9
98 suppliers
$7.00/100mg
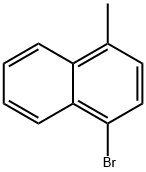
6627-78-7
115 suppliers
$13.00/1g
50672-84-9
98 suppliers
$7.00/100mg
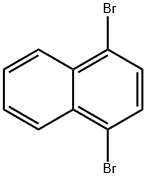
83-53-4
344 suppliers
$10.00/1g
50672-84-9
98 suppliers
$7.00/100mg