研究背景
分子催化劑的理性設(shè)計因高維結(jié)構(gòu)-性能關(guān)系(SPR)的復(fù)雜性而面臨顯著挑戰(zhàn)�����,傳統(tǒng)試錯法依賴局部修飾與經(jīng)驗參數(shù),難以突破效率瓶頸���。盡管機器學(xué)習(xí)為催化劑開發(fā)提供了數(shù)據(jù)驅(qū)動的新范式�,但其在小數(shù)據(jù)集(如新型鎳催化體系)中的應(yīng)用仍受限于模型泛化能力��。Sadphos 配體在 Pd 催化的不對稱反應(yīng)中已經(jīng)表現(xiàn)出了優(yōu)異的性能����,但在Ni催化中的應(yīng)用相對較少。浙江大學(xué)洪鑫/Zhang Shuoqing團隊基于鎳(Ni)與鈀(Pd)催化在還原消除步驟的機理相似性�����,創(chuàng)新性地提出遷移學(xué)習(xí)策略�����,利用豐富的鈀催化數(shù)據(jù)來預(yù)測鎳催化劑的配體�����,開發(fā)了一種能夠在少量實驗數(shù)據(jù)的條件下預(yù)測 Ni 催化劑性能的模型�,成功實現(xiàn)了首個鎳催化的不對稱Suzuki−Miyaura交叉偶聯(lián)反應(yīng)��。

圖1 研究背景
研究思路
研究整合了 310 條鈀催化文獻數(shù)據(jù)與低選擇性集中分布的21 條鎳 / Sadphos 實驗數(shù)據(jù),基于鈀/鎳催化共享的LM (II) Ar?中間體�����,采用 Morgan 指紋(MF)���、原子中心對稱函數(shù)(ACSF)等 5 類描述符進行機理驅(qū)動編碼����。依據(jù) MF 歐氏距離為每個 Ni/Sadphos 催化劑篩選相似鈀數(shù)據(jù)�,基于決策樹算法通過動態(tài)數(shù)據(jù)提取構(gòu)建個性化鈀模型,學(xué)習(xí)鈀催化劑的結(jié)構(gòu)-性能關(guān)系��;訓(xùn)練獨立的Delta 修正模型����,捕捉鈀→鎳選擇性偏差(SPR 偏差),對個性化鈀模型的預(yù)測結(jié)果進行修正��;最終通過整合個性化鈀模型與 Delta 修正模型的輸出��,實現(xiàn)高精度鎳催化選擇性預(yù)測(Pearson R=0.811, MAE=0.107 kcal/mol)����。
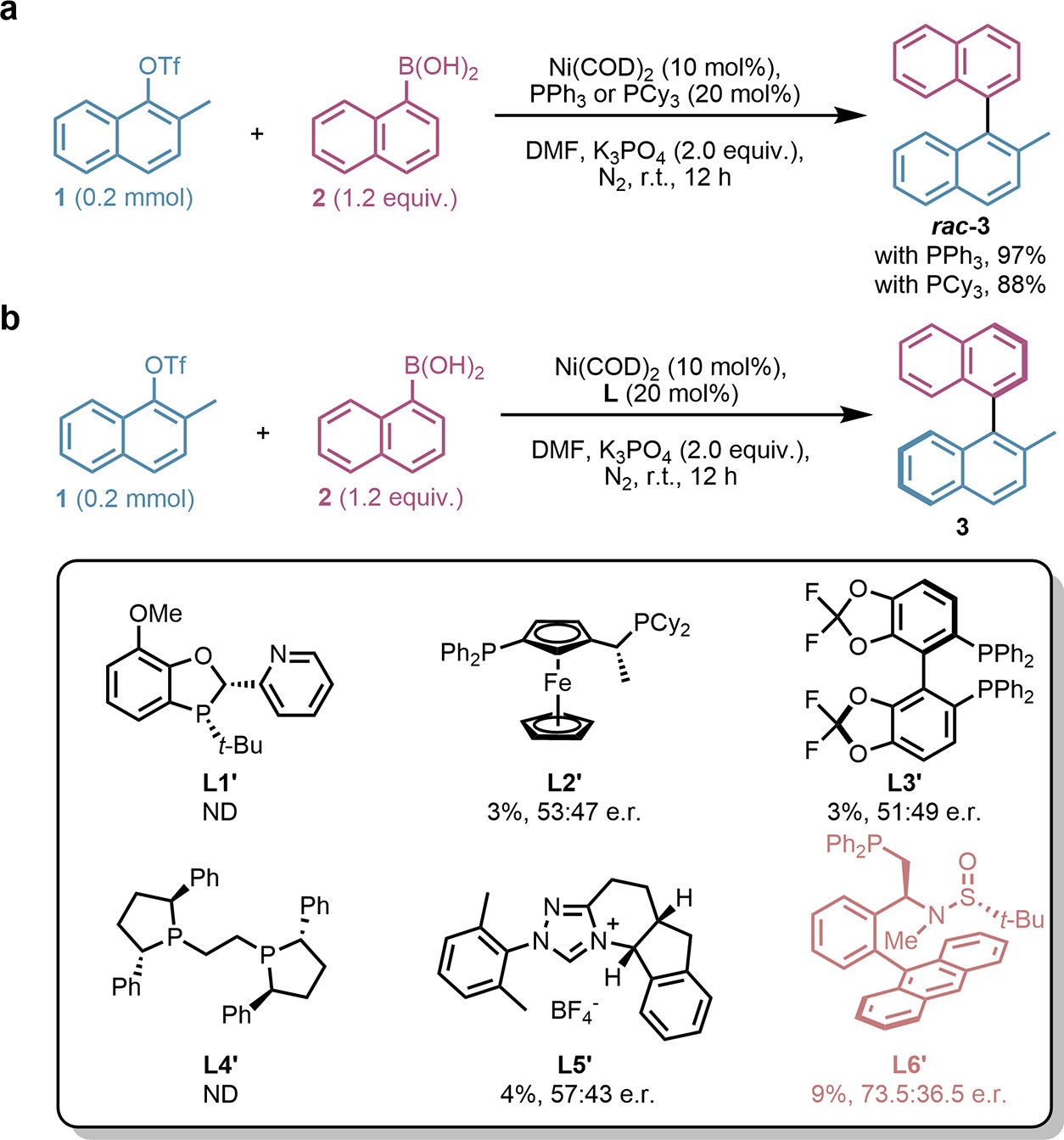
圖2 鎳催化 Suzuki-Miyaura 交叉偶聯(lián)反應(yīng)的條件優(yōu)化及手性配體篩選結(jié)果

圖3 遷移學(xué)習(xí)模型的設(shè)計策略及其建模性能
研究基于 Sadphos 的模塊化合成路線���,在 3 個可修飾位點引入多樣化官能團,構(gòu)建了包含 11284 種結(jié)構(gòu)差異化的候選配體庫��。通過整合遷移學(xué)習(xí)模型預(yù)測的對映選擇性(如 L3 預(yù)測 er = 98:2)與合成可行性評分(SAScore)�����,篩選出前3名候選配體(L1���、L2��、L3)��,其結(jié)構(gòu)兼具高預(yù)測選擇性與低合成難度����。實驗結(jié)果顯示�����,新設(shè)計的Sadphos配體L3 在鎳催化 Suzuki-Miyaura 反應(yīng)中表現(xiàn)最優(yōu)���,實現(xiàn)了96.5:3.5 er和97%產(chǎn)率���,L1和L2也展現(xiàn)較高選擇性(er = 95:5和94:6)。值得注意的是����,所有預(yù)測配體均含N-芐基取代基,是訓(xùn)練數(shù)據(jù)中未出現(xiàn)的新結(jié)構(gòu)��,體現(xiàn)了模型跨結(jié)構(gòu)空間的預(yù)測能力���。
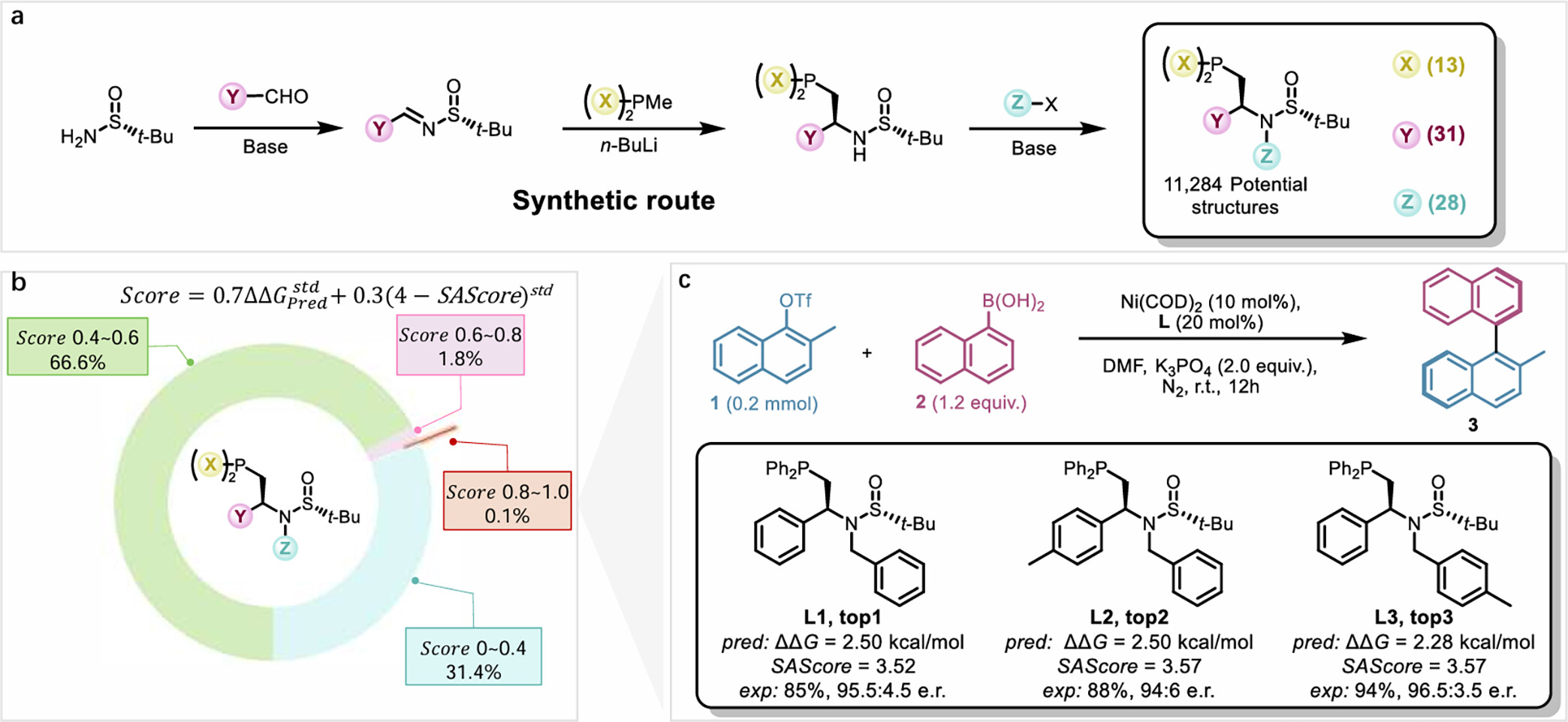
圖4 基于遷移學(xué)習(xí)模型的 Sadphos 配體設(shè)計與驗證流程
底物范圍和合成應(yīng)用
該研究進一步探討了預(yù)測得到的Ni/L3催化劑在Suzuki-Miyaura交叉偶聯(lián)反應(yīng)中的底物適用范圍及合成應(yīng)用���。實驗結(jié)果表明,該催化劑能夠兼容多種芳基硼酸����,包括給電子基團、吸電子基團和共軛體系的底物��,在含雜原子底物及親核底物的反應(yīng)中也表現(xiàn)出良好的適應(yīng)性�����。此外��,該催化劑在克級規(guī)模合成以及軸手性三萘衍生物的可控合成中,所需催化劑用量僅為1.5 mol%�����,充分體現(xiàn)了預(yù)測所得Sadphos配體L3的卓越催化效率��。

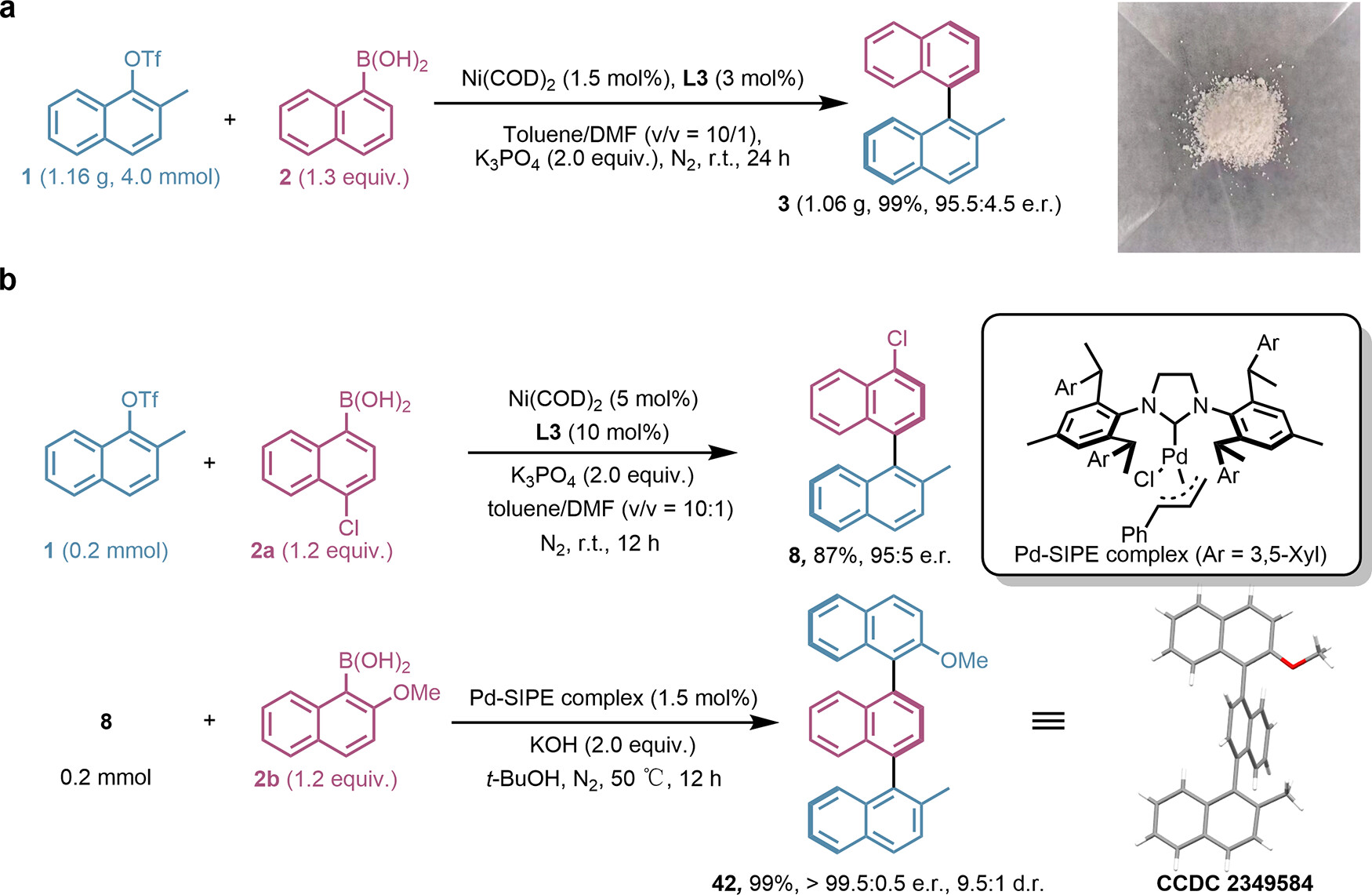
圖5 底物適用范圍及合成應(yīng)用
機理探究
密度泛函理論(DFT)計算結(jié)果表明�����,還原消除過程(TS3)既是反應(yīng)的速率決定步驟����,也是對映選擇性的決定步驟。TS3-(R)中的配體采取穩(wěn)定的六元環(huán)構(gòu)象�;而TS3-(S)中底物甲基與配體叔丁基被迫靠近,導(dǎo)致能量較高(ΔΔG‡ = 2.3 kcal/mol)��。理論計算部分的工作從分子水平驗證了鈀/鎳催化的機理相似性��,并揭示了新設(shè)計的Sadphos配體通過位阻效應(yīng)與構(gòu)象剛性實現(xiàn)高選擇性的本質(zhì)��,為跨金屬催化劑的設(shè)計提供了理論依據(jù)����。

圖6 密度泛函理論(DFT)計算
總結(jié)
本研究利用Pd和Ni催化體系在機理上的相似性,通過機器學(xué)習(xí)建立了從Pd到Ni催化的遷移學(xué)習(xí)模型����,準確預(yù)測了新型手性Sadphos配體,并成功實現(xiàn)了首例軸手性Ni催化Suzuki-Miyaura交叉偶聯(lián)反應(yīng)��。該研究為小數(shù)據(jù)量金屬催化劑的開發(fā)提供了可推廣的范式��,使配體的大規(guī)模虛擬篩選成為可能��。
文獻詳情
作者:Xin-Yuan Xu, Li-Gao Liu, Li-Cheng Xu, Shuo-Qing Zhang, Xin Hong
題目:Transfer Learning-Enabled Ligand Prediction for Ni-Catalyzed Atroposelective Suzuki−Miyaura Cross-Coupling Based on Mechanistic Similarity: Leveraging Pd Knowledge for Ni Discovery
DOI:10.1021/jacs.5c00838 (https://doi.org/10.1021/jacs.5c00838)
