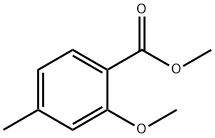
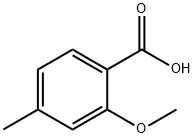
METHYL 2-METHOXY-4-METHYLBENZOATE synthesis
- Product Name:METHYL 2-METHOXY-4-METHYLBENZOATE
- CAS Number:81245-24-1
- Molecular formula:C10H12O3
- Molecular Weight:180.2
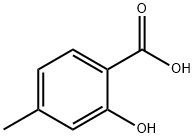
50-85-1

77-78-1
81245-24-1
1. To a 1-liter three-necked round-bottomed flask (equipped with a dropping funnel, condenser, and magnetic stirring bar) were added 4-methylsalicylic acid (50 g, 0.33 mol), K2CO3 (91.1 g, 0.66 mol, 2 eq.), and anhydrous acetone. The mixture was heated to reflux while dimethyl sulfate (112.3 g, 0.891 mol, 2.7 eq.) was added dropwise. The reaction mixture was refluxed for about 14 hours (completion of the reaction was monitored by TLC). The inorganic salt was removed by filtration and the THF filtrate was evaporated under reduced pressure to give a yellow oil. 2. The residue was dissolved in a mixture of 400 mL of methanol and concentrated NH4OH (115 mL). Stir for 30 minutes at room temperature. The methanol was removed by distillation, the residue was diluted with water (500 ml) and the oily product was extracted with ether (3 x 300 ml). The organic extracts were combined, dried with sodium sulfate, and the ether was removed under reduced pressure to give pure methyl 2-methoxy-4-methylbenzoate (57 g, 96% yield) as a yellow oil. 3. To a dry 2-liter, three-necked, round-bottomed flask (equipped with an argon inlet, a thermometer, a magnetic stirring bar, and a septum) was added THF (220 ml) and diisopropylamine (54.6 ml, 0.39 mol, 1.2 eq.). The mixture was cooled to -50 °C and a solution of 2.5 M n-BuLi (156 mL, 0.39 mol, 1.2 eq.) was slowly added through the cannula. After stirring at -50 °C for 30 min, the resulting LDA solution was cooled to -78 °C, HMPA (67.8 mL, 0.39 mol, 1.2 eq.) was added, followed by a solution of methyl 2-methoxy-4-methylbenzoate (57 g, 0.32 mol) in THF (220 mL), keeping the temperature at -78 °C. The reaction mixture was stirred at -78 °C for an additional 2 h and then transferred to a flask containing 100 g of dry ice and THF (200 mL). After stirring at -78 °C for 30 min, the mixture was slowly warmed to room temperature and poured into water (1.5 liters). The organic layer was separated and the aqueous layer was further extracted with ether (3 x 1 liter). The aqueous layer was acidified with 10% H2SO4 (150 ml) and the reaction product was extracted with CH2Cl2 (3 x 1.5 liters). The organic extracts were combined, dried with anhydrous Na2SO4, the solvent was removed under reduced pressure, and the crude product was purified by fast chromatography (eluent: EtOAc:hexane 3:7) and recrystallized from CH2Cl2:hexane 1:1 to give pure methyl 4-carboxymethyl-2-methoxybenzoate (38.8 g, 54.7% yield). 4. Methyl 4-carboxymethyl-2-methoxybenzoate (12.7 g, 0.0567 mol) was dissolved in glacial acetic acid (100 mL) and bromine (3.2 mL, 0.0624 mol, 1.1 eq.) was added dropwise via a syringe, maintaining the internal temperature at room temperature. After stirring for 4 h, the AcOH was removed by evaporation and the orange oil was evaporated twice with toluene to give an orange solid. The substance was ground with CH2Cl2 and filtered to remove some of the precipitated product. The remaining product in the filtrate was concentrated on a silica gel column, eluting with EtOAc:hexane (6:4) containing 1% AcOH. Further purification by a second chromatography and recrystallization from EtOAc:hexane gave pure methyl 5-bromo-4-carboxymethyl-2-methoxybenzoate (9 g total, 52% yield) as a white solid.
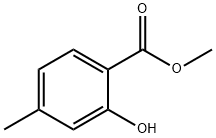
Yield: 99%
Reaction Conditions:
with sodium hydride in N,N-dimethyl-formamide at 0; for 1 h;
Steps:
33.1 Step one, compound 2
Compound 1 (7.6 g, 50 mmol) was dissolved in N,N-dimethylformamide (100 mL), and methyl iodide (21.3 g, 150 mmol) was added.After the reaction system was cooled to 0°C, sodium hydride (60%, 6g, 150mmol) was slowly added in batches, stirred for 1 hour, and water (20mL) was added to quench the reaction.The solution obtained above was extracted with ethyl acetate (3x 200mL), the organic phase was washed successively with water (2x 200mL), saturated sodium chloride solution (200mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain compound 2 (yellow oily) Liquid, 9.0g, yield 99%).
References:
CN112939961, 2021, A Location in patent:Paragraph 0424-0428
4670-56-8
179 suppliers
$10.00/1g

74-88-4
356 suppliers
$15.00/10g
81245-24-1
126 suppliers
$42.00/1g
704-45-0
133 suppliers
$14.00/1g
74-88-4
356 suppliers
$15.00/10g
81245-24-1
126 suppliers
$42.00/1g