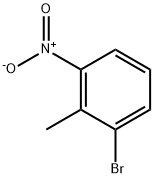
2-Bromo-6-nitrotoluene synthesis
- Product Name:2-Bromo-6-nitrotoluene
- CAS Number:55289-35-5
- Molecular formula:C7H6BrNO2
- Molecular Weight:216.03
603-83-8
55289-35-5
1. Suspend 2-methyl-3-nitroaniline (45.6 g, 0.3 mol) in water (200 mL) and 40% aqueous hydrobromic acid (100 mL) and heat to reflux for 10 minutes. Cool to 0°C and slowly add aqueous sodium nitrite (20.7 g, 0.3 mol) (100 mL) dropwise, controlling the temperature to no more than 5°C. Continue stirring at 0-5°C for 30 min. 2. Slowly add the above diazonium salt solution to a stirred mixture of cuprous bromide (43.1 g, 0.3 mol) in hydrobromic acid (150 mL) and water (150 mL), and after stirring at room temperature for 30 minutes, heat the reaction to 70°C for 1 hour. The reaction solution was poured into ice water and extracted with dichloromethane (400 mL x 3), the organic phases were combined, washed sequentially with saturated aqueous sodium bicarbonate and brine, dried over anhydrous magnesium sulfate, filtered and concentrated. 3. The residue was purified by column chromatography (petroleum ether as eluent) to afford 1-bromo-2-methyl-3-nitrobenzene (58 g, 89% yield) as a light yellow solid. 4. 1-Bromo-2-methyl-3-nitrobenzene (37.0 g, 0.17 mol), N-bromosuccinimide (61 g, 0.34 mol) and azobisisobutyronitrile (0.7 g, 4.2 mmol) were refluxed in carbon tetrachloride (500 mL) overnight. The filtrate was filtered and concentrated to give the red liquid crude product 1-bromo-2-(bromomethyl)-3-nitrobenzene (53 g), which was used directly in the next step. 5. The crude product 1-bromo-2-(bromomethyl)-3-nitrobenzene (53 g) was stirred with a solution of sodium acetate (32.8 g, 0.4 mol) in N,N-dimethylformamide (300 mL) at 70 °C overnight. The reaction solution was diluted with water, extracted with ethyl acetate, the organic phases were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. 6. The residue was purified by column chromatography (petroleum ether/ethyl acetate=20/1) to give white solid 2-bromo-6-nitrobenzyl acetate (24.0 g, two-step overall yield 51%). 7. Under nitrogen protection, nitrogen was bubbled into a solution of 2-bromo-6-nitrobenzyl acetate (24.5 g, 0.089 mol) in 1,4-dioxane (500 mL) for 20 min, potassium acetate (38.4 g, 0.358 mol), [1,1'-bis(diphenylphosphino)ferrocene]palladium dichloride (3.65 g, 4.5 mmol) and bis(pinacolato) alcoholato)diboron (34.1 g, 0.13 mol) and stirred at 95 °C for 20 hours. After cooling and concentration under reduced pressure, the residue was partitioned with ethyl acetate and water, the organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. 8. The residue was purified by column chromatography (petroleum ether/ethyl acetate=20/1) to afford 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-nitrobenzenemethanol as a white solid (11.0 g, 38% yield). lC-MS (ESI) m/z 322 [M+1]+ (calculated value 321.1). 9. To a solution of 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-nitrobenzenemethanol (11.0 g, 0.034 mol) in methanol (350 mL) was added an aqueous solution of 5N sodium hydroxide (17 mL, 0.085 mol) and refluxed under nitrogen protection for 24 hours. Concentrate under reduced pressure and dissolve in tetrahydrofuran (200 mL), add 5N hydrochloric acid (60 mL, 0.3 mol) and stir at 40°C for 16 hours. After cooling, it was diluted with ethyl acetate and poured into brine, the organic phase was separated, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. 10. The residue was recrystallized in a mixed solvent of ethyl acetate and petroleum ether to give 4-nitrobenzo[c][1,2]oxaborol-1(3H)-ol as a yellow solid (5.0 g, 81% yield).LC-MS (ESI) m/z 180 [M+1]+ (calculated value 179.0).1H NMR (300MHz, DMSO-d6): δ 9.59 ( s, 1H), 8.33 (d, 1H), 8.14 (d, 1H), 7.71 (t, 1H), 5.37 (s, 2H). 11. A solution of 4-nitrobenzo[c][1,2]oxaborol-1(3H)-ol (2.7 g, 0.015 mol) in methanol (150 mL) was hydrogenated by addition of palladium/carbon (1.0 g) overnight at atmospheric pressure and room temperature to afford the target product, 2-bromo-6-nitrotoluene (1.5 g, 66% yield). lc-MS (ESI) m/z 150 [M+1 ]+ (calculated value 149.1).1H NMR (300MHz, DMSO-d6): δ 8.92 (s, 1H), 7.02 (t, 1H), 6.90 (d, 1H), 6.62 (d, 1H), 4.99 (s, 2H), 4.77 (s, 2H).
603-83-8
301 suppliers
$11.00/5g
55289-35-5
304 suppliers
$13.00/1g
Yield:55289-35-5 89%
Reaction Conditions:
Stage #1:3-nitro-o-tolylamine with hydrogen bromide in water for 0.166667 h;Reflux;
Stage #2: with sodium nitrite in water at 0 - 5; for 0.5 h;
Stage #3: with copper(I) bromide in water at 20 - 70; for 1.5 h;
Steps:
Preparation of benzoxaborole 2f
2-Methyl-3-nitrobenzenamine (45.6 g, 0.3 mol) was suspended in water (200 mL) and HBr (100 mL, 40% aq.) and the mixture was heated to reflux for 10 min. Then the mixture was cooled to 0 °C and NaNO2 (20.7 g, 0.3 mol) in water (100 mL) was added dropwise at such a rate that the temperature did not exceed 5 °C. The diazonium solution was stirred for a further 30 min at 0-5 °C and then added slowly to a stirred mixture of CuBr (43.1 g, 0.3 mol) in HBr (150 mL) and water (150 mL) at room temperature. The mixture was stirred at room temperature for 30 min and then heated to 70°C for 1h. The mixture was poured onto ice and extracted with CH2Cl2 (400 mL×3), and washed with sat. NaHCO3 and brine, dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography with petroleum ether as eluent to give a pale yellow solid (58 g, 89%)The mixture of 1-bromo-2-methyl-3-nitrobenzene (37.0 g, 0.17 mol), NBS (61 g, 0.34 mol) and AIBN (0.7 g, 4.2 mmol) in CCl4 (500 mL) was refluxed overnight. The mixture was filtered and the filtrate was concentrated to give a red liquid (53 g) as a crude product which was used in the next step without any purification.1-Bromo-2-(bromomethyl)-3-nitrobenzene (53 g) and NaOAc (32.8 g, 0.4 mol) in DMF (300 mL) was stirred at 70 °C overnight. The mixture was then diluted with water and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography with petroleum ether/ethyl acetate = 2 0/1 eluent to give a white solid (24.0 g, 51% over two steps).To 2-bromo-6-nitrobenzyl acetate (24.5 g, 0.089 mol) in 1,4-Dioxane (500 mL) was bubbled nitrogen in the solution for 20 min. Potassium acetate (38.4 g, 0.358mol), Pd(dppf)Cl2 (3.65 g, 4.5 mmol) and bis(pinacolato)diboron (34.1 g, 0.13 mol) were added and the reaction mixture was stirred under nitrogen at 95 °C for 20 h. The reaction mixture was then cooled and was evaporated under vacuum. The residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography with petroleum ether/ethyl acetate = 20/1 as eluent to give a white solid (11.0 g, 38%). LC-MS (ESI) m/z 322 [M+1]+, (calcd MS 321.1).To a solution of 6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-nitrobenzyl acetate (11.0 g, 0.034 mol) in methanol (350 mL) was added 5 N NaOH (17 mL, 0.085 mol). The reaction mixture was stirred and refluxed under nitrogen for 24h. The reaction mixture was then concentrated under vacuum and was dissolved in 200 mL of tetrahydrofuran (THF). HCl (5 N, 60 mL, 0.3 mol) was added and the reaction mixture was stirred and heated at 40 °C for 16h. The reaction mixture was cooled, diluted with EtOAc and poured into brine. The separated organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was recrystallized in the mixture of ethyl acetate and petroleum ether to give a yellow solid (5.0 g, 81%). LC-MS (ESI) m/z 180 [M+1]+, (calcd MS 179.0). 1H NMR (300 MHz, DMSO-d6): δ 9.59 (s, 1H), 8.33 (d, 1H), 8.14 (d, 1H), 7.71 (t, 1H), 5.37 (s, 2H).4-Nitrobenzo[c][1,2]oxaborol-1(3H)-ol (2.7 g, 0.015 mol) in methanol (150 mL) was added Pd/C (1.0 g) and the hydrogenation was conducted at one atmosphere and room temperature overnight to provide the desired product 2f as a yellow solid (1.5 g, 66%). LC-MS (ESI) m/z 150 [M+1]+, (calcd MS 149.1). 1H NMR (300 MHz, DMSO-d6): δ 8.92 (s, 1H), 7.02 (t, 1H), 6.90 (d, 1H), 6.62 (d, 1H), 4.99(s, 2H), 4.77(s, 2H).
References:
Li, Xianfeng;Zhang, Suoming;Zhang, Yong-Kang;Liu, Yang;Ding, Charles Z.;Zhou, Yasheen;Plattner, Jacob J.;Baker, Stephen J.;Bu, Wei;Liu, Liang;Kazmierski, Wieslaw M.;Duan, Maosheng;Grimes, Richard M.;Wright, Lois L.;Smith, Gary K.;Jarvest, Richard L.;Ji, Jing-Jing;Cooper, Joel P.;Tallant, Matthew D.;Crosby, Renae M.;Creech, Katrina;Ni, Zhi-Jie;Zou, Wuxin;Wright, Jon [Bioorganic and Medicinal Chemistry Letters,2011,vol. 21,# 7,p. 2048 - 2054] Location in patent:supporting information; experimental part
95-46-5
306 suppliers
$10.00/10 g
55289-35-5
304 suppliers
$13.00/1g

606-20-2
0 suppliers
$13.00/5g
55289-35-5
304 suppliers
$13.00/1g
88-72-2
271 suppliers
$15.00/25g
60956-26-5
300 suppliers
$8.00/10g
55289-35-5
304 suppliers
$13.00/1g
95-46-5
306 suppliers
$10.00/10 g
7149-70-4
188 suppliers
$9.00/1g

7745-93-9
294 suppliers
$5.00/5g

41085-43-2
200 suppliers
$6.00/1g
55289-35-5
304 suppliers
$13.00/1g