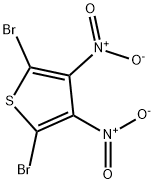
2,5-DIBROMO-3,4-DINITROTHIOPHENE synthesis
- Product Name:2,5-DIBROMO-3,4-DINITROTHIOPHENE
- CAS Number:52431-30-8
- Molecular formula:C4Br2N2O4S
- Molecular Weight:331.93
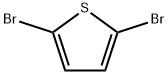
3141-27-3
52431-30-8
GENERAL STEPS: Preparation The synthesis of 2,5-dibromo-3,4-dinitrothiophene from 2,5-dibromothiophene in Example 1 was carried out as follows: firstly, a mixed acid was prepared by mixing 11 mL of fuming nitric acid with 20 mL of fuming sulfuric acid. Subsequently, 13 mL of concentrated sulfuric acid was added to this mixed acid to prepare a reaction solution. To this solution, 7.5 g (31 mmol) of 2,5-dibromothiophene was slowly added dropwise while maintaining the reaction temperature between 20 and 30 °C by means of a water bath with continuous stirring for 3 hours. Upon completion of the reaction, the reaction solution was transferred to a vessel containing 90 g of ice to terminate the reaction. The resulting solid product was collected by filtration and purified by recrystallization with methanol to give the final target product 2,5-dibromo-3,4-dinitrothiophene. The yield based on 2,5-dibromothiophene was 66%.
3141-27-3
319 suppliers
$5.00/10g
52431-30-8
194 suppliers
$24.00/1g
Yield:52431-30-8 88%
Reaction Conditions:
with sulfuric acid;HNO3 in lithium hydroxide monohydrate at 20 - 30; for 4 h;Cooling with ice;
Steps:
2 2,5-dibromo-3,4-dinitrothiophene
Compound 5 was prepared according to the procedure reported (Mozingo R, Harris SA, Wolf DE, Hoffhine CE, Easton, NR, Folkers K, et al. Hydrogenation of Compounds Containing Divalent Sulfur. J Am Chem Soc 1945; 67: 2092-5, Hailu H , Atsbeha B, Admassie S, Mammo W, Raju VJT, Chebude Y, et al. Variable denticity of a multidentate terthiophene derivative towards ni (ii) and zn (ii) structural studies. Bull Chem Soc Ethiop 2011; 25: 221-31 ] To a solution of exothermic H2SO4 (110 mL),Exothermic HNO3 (60 mL) and concentratedH2SO4 (73 mL) was mixed in a flask,Cooled in an ice bath.2,5-dibromothiophene 4(2,5-dibromothiophene, 21.54 g, 89 mmol) was added dropwise and the temperature was maintained at 20-30 ° C. The reaction was stirred in ice for 4 h. The solid residue was filtered and washed with water and then purified by column chromatography (ethyl acetate in 20% hexane) to obtain light yellow powder (25.94 g, 88%).
References:
KR2018/17709,2018,A Location in patent:Paragraph 0173; 0176; 0180-0182

188290-36-0
1 suppliers
inquiry
52431-30-8
194 suppliers
$24.00/1g
3141-24-0
254 suppliers
$7.00/5g
52431-30-8
194 suppliers
$24.00/1g
