
*Storage: {[sel_prStorage]}
*Shipping: {[sel_prShipping]}
4.5

 *For research use only!
*For research use only!
Change View
| Size | Price | VIP Price | USA Stock *0-1 Day |
Global Stock *5-7 Days |
In Stock | ||
| {[ item.pr_size ]} |
Inquiry
{[ getRatePrice(item.pr_usd, 1,1,item.pr_is_large_size_no_price, item.pr_usd) ]} {[ getRatePrice(item.pr_usd,item.pr_rate,1,item.pr_is_large_size_no_price, item.discount_usd) ]} {[ getRatePrice(item.pr_usd, 1,1,item.pr_is_large_size_no_price, item.pr_usd) ]} |
Inquiry {[ getRatePrice(item.pr_usd,item.pr_rate,item.mem_rate,item.pr_is_large_size_no_price, item.vip_usd) ]} | Inquiry {[ item.pr_usastock ]} In Stock Inquiry - | {[ item.pr_chinastock ]} {[ item.pr_remark ]} In Stock 1-2 weeks - Inquiry - | Login | - + | Inquiry |
Please Login or Create an Account to: See VIP prices and availability
1-2weeks
Inquiry
{[ getRatePrice(item.pr_usd,item.pr_rate,item.mem_rate,item.pr_is_large_size_no_price, item.vip_usd) ]}
{[ getRatePrice(item.pr_usd, 1,1,item.pr_is_large_size_no_price, item.pr_usd) ]}
{[ getRatePrice(item.pr_usd,1,item.mem_rate,item.pr_is_large_size_no_price, item.pr_usd) ]}
Inquiry
{[ getRatePrice(item.pr_usd,item.pr_rate,1,item.pr_is_large_size_no_price, item.vip_usd) ]}
{[ getRatePrice(item.pr_usd, 1,1,item.pr_is_large_size_no_price, item.pr_usd) ]}
{[ getRatePrice(item.pr_usd, 1,1,item.pr_is_large_size_no_price, item.pr_usd) ]}
In Stock
- +
Please Login or Create an Account to: See VIP prices and availability
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.

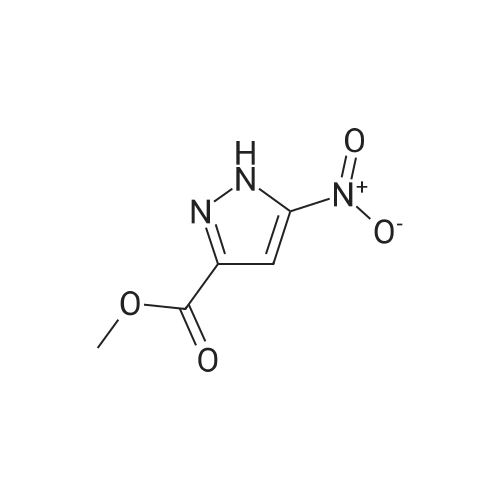
| CAS No. : | 181585-93-3 |
| Formula : | C5H5N3O4 |
| M.W : | 171.11 |
| MDL No. : | MFCD08236713 |
| InChI Key : | OTINMTPELZSAPX-UHFFFAOYSA-N |
| Pubchem ID : | 135452337 |
| GHS Pictogram: |

|
| Signal Word: | Warning |
| Hazard Statements: | H302-H315-H319-H332-H335 |
| Precautionary Statements: | P261-P280-P305+P351+P338 |
| Num. heavy atoms | 12 |
| Num. arom. heavy atoms | 5 |
| Fraction Csp3 | 0.2 |
| Num. rotatable bonds | 3 |
| Num. H-bond acceptors | 5.0 |
| Num. H-bond donors | 1.0 |
| Molar Refractivity | 38.69 |
| TPSA ? Topological Polar Surface Area: Calculated from |
100.8 ?2 |
| Log Po/w (iLOGP)? iLOGP: in-house physics-based method implemented from |
0.44 |
| Log Po/w (XLOGP3)? XLOGP3: Atomistic and knowledge-based method calculated by |
0.56 |
| Log Po/w (WLOGP)? WLOGP: Atomistic method implemented from |
0.1 |
| Log Po/w (MLOGP)? MLOGP: Topological method implemented from |
-0.56 |
| Log Po/w (SILICOS-IT)? SILICOS-IT: Hybrid fragmental/topological method calculated by |
-1.23 |
| Consensus Log Po/w? Consensus Log Po/w: Average of all five predictions |
-0.14 |
| Log S (ESOL):? ESOL: Topological method implemented from |
-1.36 |
| Solubility | 7.4 mg/ml ; 0.0432 mol/l |
| Class? Solubility class: Log S scale |
Very soluble |
| Log S (Ali)? Ali: Topological method implemented from |
-2.25 |
| Solubility | 0.964 mg/ml ; 0.00563 mol/l |
| Class? Solubility class: Log S scale |
Soluble |
| Log S (SILICOS-IT)? SILICOS-IT: Fragmental method calculated by |
-0.69 |
| Solubility | 35.1 mg/ml ; 0.205 mol/l |
| Class? Solubility class: Log S scale |
Soluble |
| GI absorption? Gatrointestinal absorption: according to the white of the BOILED-Egg |
High |
| BBB permeant? BBB permeation: according to the yolk of the BOILED-Egg |
No |
| P-gp substrate? P-glycoprotein substrate: SVM model built on 1033 molecules (training set) |
No |
| CYP1A2 inhibitor? Cytochrome P450 1A2 inhibitor: SVM model built on 9145 molecules (training set) |
No |
| CYP2C19 inhibitor? Cytochrome P450 2C19 inhibitor: SVM model built on 9272 molecules (training set) |
No |
| CYP2C9 inhibitor? Cytochrome P450 2C9 inhibitor: SVM model built on 5940 molecules (training set) |
No |
| CYP2D6 inhibitor? Cytochrome P450 2D6 inhibitor: SVM model built on 3664 molecules (training set) |
No |
| CYP3A4 inhibitor? Cytochrome P450 3A4 inhibitor: SVM model built on 7518 molecules (training set) |
No |
| Log Kp (skin permeation)? Skin permeation: QSPR model implemented from |
-6.95 cm/s |
| Lipinski? Lipinski (Pfizer) filter: implemented from |
0.0 |
| Ghose? Ghose filter: implemented from |
None |
| Veber? Veber (GSK) filter: implemented from |
0.0 |
| Egan? Egan (Pharmacia) filter: implemented from |
0.0 |
| Muegge? Muegge (Bayer) filter: implemented from |
1.0 |
| Bioavailability Score? Abbott Bioavailability Score: Probability of F > 10% in rat |
0.55 |
| PAINS? Pan Assay Interference Structures: implemented from |
0.0 alert |
| Brenk? Structural Alert: implemented from |
2.0 alert: heavy_metal |
| Leadlikeness? Leadlikeness: implemented from |
No; 1 violation:MW<1.0 |
| Synthetic accessibility? Synthetic accessibility score: from 1 (very easy) to 10 (very difficult) |
2.26 |
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.

 [ 67-56-1 ]
[ 67-56-1 ]
 [ 181585-93-3 ]
[ 181585-93-3 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 100% | With thionyl chloride; N,N-dimethyl-formamide; at 0 - 70℃; for 4h; | Step a. A solution of 3-nitro-lH-pyrazole-5-carboxylic acid (63.66 mmol) in MeOH (100 ml) and catalytic amount of DMF (0.05 ml) was stirred at 0C for 10 min. SOCl2 (165.52 mmol) was added dropwise to the reaction mixture at 0C. The reaction mixture was allowed to warm up to rt and then heated at 70C for 4 h. The resulting reaction mixture was concentrated under reduced pressure. The obtained residue was azeotropically distilled using MeOH (100 ml) yielding methyl 3-nitro-lH-pyrazole-5-carboxylate (quantitative). MS: ES- 170.12. |
| 95% | With thionyl chloride; at 0℃; for 2h;Reflux; | To a solution of 5-nitro-1H-pyrazole-3-carboxylic acid (1.13 g, 7.19 mmol) in anhydrous methanol (20 ml) at to 0C was added thionyl chloride (2.23 g, 1.37 ml, 18.7 mmol) dropwise. The resulting solution was heated to reflux for 2 h. The cooled solution was evaporated to dryness to give 5-nitro-2H-pyrazole-3-carboxylic acid methyl ester (1.17 g, 95%) as a white solid. |
| 95% | With sulfuric acid; at 65℃; | [0564] To a solution of 5-nitro-2H-pyrazole-3-carboxylic acid (1.00 g, 6.37 mmol) in MeOH (20 mL) was added cone. H2SO4 (2 mL), and the resulting mixture was stirred at 65 C overnight. Then MeOH was removed in vacuum to give a yellow residue, which was purified by silica gel column with DCM as eluent to afford 5-nitro-2H-pyrazole-3-carboxylic acid methyl ester (1.04 g, yield: 95%) as a white solid. [0565] 1H NMR (400 MHz, CDC13): delta = 11.63 (brs, 1H), 7.41 (s, 1H), 4.01 (s, 3H). |
| 95% | With thionyl chloride; at 80℃; for 16h; | To a stirred solution of 3-nitro-lH-pyrazole-5-carboxylic acid(A58, 25.0 g, 159.14 mmol) in Methanol(250 mL) was added dropwise SOCk(23.09 mL, 318.28 mmol) mL). The reaction mixture was stirred for 16 h at 80C. After completion, addition of n-hexane(500 mL) solid precipitated, filtered and dried under reduced pressure to afford methyl 3-nitro-lH-pyrazole-5-carboxylate A59 as a white solid. Yield: 26.0 g(95%) LC-MS(ES) m/z : 170.14[M+H]+. |
| 93% | With sulfuric acid; for 5h;Reflux; | Commercial 3-nitro-1H-pyrazole-5-carboxylic acid (99.6%purity, Aldrich) (1.54 g, 10mM)was dissolved in methanol (15 mL), and 95%H2SO4 (0.14 mL) was added.The mixture was refluxed for 5 h. The solvent was evaporated under reduced pressure. A precipitatewas dissolved in ethyl acetate and washed with 5% NaHCO3 (2 x10 mL) and water (1 x10 mL).The organic layer was dried with MgSO4, and the ethyl acetate was evaporated. The residue wasdissolved in dichloromethane and purified on a silica-gel column (60H Merck 1.05553) eluted withDCM:MeOH, from 2%±6% of MeOH; crystallization from a mixture of diethyl ether/hexane. Yield:1.28 g, 93%. 1H NMR (400MHz, DMSO-d6) delta(ppm): 15.25 (1H, s, N-HPz), 7.54 (1H, s, C-HPz), 3.91 (3H,s, CH3) (Figure S19). Melting point by DSC: 148 C (Figure S24). TLC CHCl3/MeOH/AcOH (90:8:2)Rf = 0.74. |
| 89.3% | With thionyl chloride; at 0℃; for 2h;Reflux; | Step 11: Preparation of 5-Nitro-2H-pyrazole-3-carboxylic acid methyl ester A lOmL single-neck round-bottomed flask was charged with 5-nitro-lH-pyrazole-3-carboxylic acid (4.0 g, 25.5 mmol) and anhydrous MeOH (40.0 ml). The reaction mixture was cooled to 0 C in an ice/water cooling bath. To this mixture, thionyl chloride (7.88 g, 4.83 ml, 66.2 mmol) was added dropwise. After the addition was complete, the bath was removed and the reaction mixture was heated at reflux for 2 h. The reaction mixture was then concentrated to dryness under reduced pressure to give the desired product (4.36 g, 89.3%). (M+Na)+ = 198.9 m/e |
| 81% | To a solution of 3-nitro-1H-pyrazole-5-carboxylic acid (2.00 g, 12.73 mmol) in methanol (80 mL) was added thionyl chloride (1.00 ml_, 13.70 mmol). The reaction mixture was heated to reflux for 18 hours. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate and washed with saturated sodium bicarbonate, water and brine. The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated in vacuo to afford methyl 3-nitro-1H- pyrazole-5-carboxylate in 81 % yield (1.77 g): 1H NMR (300 MHz, CDCI3) delta 9.97 (br s, 1 H), 7.39 (s, 1H), 3.99 (s, 3H); MS (ES+) m/z 171.0 (M). | |
| 81% | With thionyl chloride; at 0℃; for 2.33333h;Reflux; | To a sobr& of 3-nibD-lH-pa7de-5-ca1boxy& acid (69.75 g, 444 intol) (ArkPhant in MDH (1L)was ad&dthirckhide (S4 nL, llS4numl) O C. Th nuetirewas sth1 £Srabciat2J ittat about 0 C then Inted to D?fbIx fc about 2k. Th iemlbrg sobxon was oDirentated urderpressuie to give fl1 (61.25 g, Si %): LCIMS (Table1, Metlvd I) R = 1.42 ntha.; MS m&: 169 (M-i-H). |
| 74% | With thionyl chloride; at 0 - 70℃; for 4h; | To a stirred solution of 3-nitro-1H-pyrazole-5-carboxylic acid 19 (2.0 g,11.69 mmol)) in methanol (50 mL), thionyl chloride (2.07 g, 17.54 mmol) was charged at0C, in portions. The reaction mixture was heated at 70C for 4 hour. The reaction p1ixture was evaporated and basified with saturated sodium bicarbonate solution to pH 9. The aqueous layer was extracted with dichloromethane (2 x 100 mL). The combined organic extract was washed with water (3 x 30 mL). The organic layer was dried (Na2SO4), filtered and evaporated to obtain methyl 3-nitro-1H-pyrazole-5-carboxylate 20 (1.5 g, 74% yield) as brown sticky solid. MS: 171.03 (M+H). |
| 70% | With sulfuric acid; at 20 - 65℃; for 89h; | Step 1: Methyl 3-nitro-lH-pyrazole-5-carboxylate [00198] A stirred solution of 3-nitro-lH-pyrazole-5-carboxylic acid (940 mg, 5.98 mmol) in MeOH (10 mL) at 20 C was treated cautiously with cone. H2S04 (1 mL). The reaction mixture was heated to 65 C for 89 h, then concentrated to dryness. The resulting white suspension was triturated with water (10 mL) and the resulting precipitate collected by filtration to yield the title compound as a white solid (718 mg, 70%). LCMS (ES+) 172 (M+H)+. |
| 69% | With thionyl chloride; at 0℃; for 16h;Reflux; | 5.1.45 Methyl 3-nitro-1H-pyrazole-5-carboxylate (9) To a solution of 8 (20.0 g, 127.32 mmol) in anhydrous methanol (70 mL) was added SOCl2 (10.2 mL, 140.11 mmol) dropwise at 0 C. The resulting mixture was heated at reflux for 16 h, and then concentrated in vacuo. The residue was dissolved in ethyl acetate (200 mL) and washed with saturated NaHCO3 solution (40 mL * 2), water (40 mL), and brine (40 mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the residue was crystallized from ethyl acetate/hexane to afford 9 as an off-white solid (15.0 g, 69%). 1H NMR (300 MHz, CDCl3) delta 11.52 (br s, 1H), 7.41 (s, 1H), 4.01 (s, 3H); MS (ES-) m/z 170.0 (M-1). |
| 13.8% | Step I - Preparation of5-nitro-2H-pyrazole-3-carboxylic acid methyl ester (525):; [0136] To 5-nitro-2H-pyrazole-3-carboxylic acid (524, 10.0 g, 0.0637 mol) in methanol (100.0 inL) was added concentrated sulfuric acid (1.00 mL, 0.0180 mol). The reaction was stirred at room temperature overnight. The reaction was poured into aqueous potassium carbonate and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% ethyl acetate in hexane to give a white solid (525, 1.5 g, 13.8%). | |
| With sulfuric acid; for 20h;Heating / reflux; | delta-Nitro^H-pyrazole-S-carboxylic acid methyl ester: A solution of delta-Nitro^H-pyrazole-S-carboxylic acid (5 gm, 32 mmol) in100 ml of 2% sulfuric acid methanol was refluxed for 20 hours. The mixture was cooled to room temperature and added to saturated sodium bicarbonate. The mixture was extracted with ethyl acetate three times to obtain 3.59 gm of title product after drying over magnesium sulfate, filtering and evaporating to dryness. ESI M+1 =171. | |
| With sulfuric acid; at 65℃; | To a solution of 3-nitro-JH-pyrazole-5-carboxylic acid (1 57 g, 10.0 mmol) in MeOH(20 mL) was added conc.fl2S04 (2.0 mL). The resulting mixture was stirred at 65 C overnight.Then the mixture was concentrated in vacuo to gtve a residue, which was purified by silica gelcolumn (DCM/J1eOH '" 50/l) to afford methyl 3-nitro-1 H-pyrazole-5-carboxy late (1.42 g, yield:83?--o) as yellow solid. |

[ 2746-25-0 ]
[ 181585-93-3 ] [ 625386-10-9 ]
[ 625386-10-9 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 99% | With potassium carbonate; In tetrahydrofuran; at 20℃; for 2h; | To a suspension of methyl 3-nitro-1 H-pyrazole-5-carboxylate (1 ,77 g, 10.35 mmol) and potassium carbonate (2.06 g, 14.91 mmol) in tetrahydrofuran (100 mL) was added 4-methoxy benzyl bromide (1.58 mL, 10.93 mmol). The reaction mixture was stirred for 2 hours at ambient temperature and filtered. The filtrate was concentrated in vacuo to afford methyl 1-(4-methoxybenzyl)-3-nitro-1H-pyrazole-5-carboxylate in 99% yield (3.00 g): 1H NMR (300 MHz, CDCI3) 6 7.37-7.32 (m, 3H), 6.86-6.81 (m, 2H), 5.75 (s, 2H), 3.90 (s, 3H), 3.76 (s, 3H); MS (ES+) m/z 292.2 (M + 1). |
| With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 5h; | To a mixture of 9 (13.00g, 76.02mmol) and K2CO3 (18.30g, 132.40mmol) in anhydrous N,N-dimethylformamide (350mL) was added 4-methoxybenzyl bromide (15.0mL, 120.38mmol). The resulting mixture was heated at 60C for 5h. The solid was filtered off and the filtrate was concentrated under reduced pressure at a temperature below 80C. The residue was dissolved in ethyl acetate (500mL), washed with 10% aqueous NH4Cl solution (100mL×2) and brine (100mL), dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the residue was crystallized from methanol to afford crude 10 as an off-white solid (23.0g), which was used in the next step without further purification. For an analytical sample, a small amount of crude material was purified by column chromatography eluted with a gradient of 50-80% dichloromethane in hexanes to afford 10 as an off-white solid. 1H NMR (300MHz, CDCl3) delta 7.39 (s, 1H), 7.34 (d, J=8.6Hz, 2H), 6.85 (d, J=8.6Hz, 2H), 5.76 (s, 2H), 3.92 (s, 3H), 3.78 (s, 3H) |
[ 181585-93-3 ]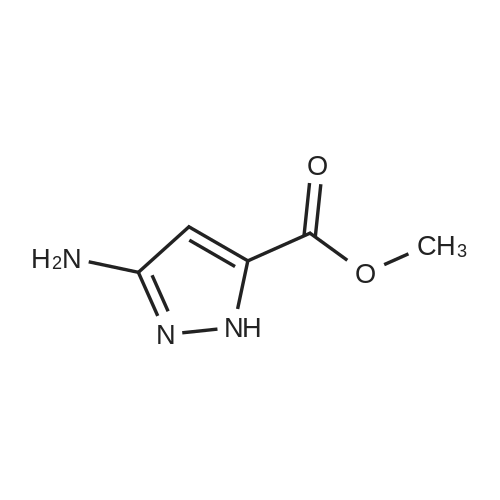 [ 1312929-42-2 ]
[ 1312929-42-2 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 100% | With hydrogen;palladium 10% on activated carbon; In methanol; under 2585.81 Torr; for 24h; | To a solution of methyl 3-nitro-lH-pyrazole-5-carboxylate (14A) (5 g, 29.22 mmol) in methanol (75 mL) was added Pd/C (10% on C, 0.6 g). The resulting mixture was hydrogenated at 50 psi for 24 h. After filtering the catalyst through a pad of Celite, the filtrate wasconcentrated in vacuum to dryness and the residue was purified by flash columnchromatography (silica gel, eluting with MeOH/CHCl3 0 to 20%) to furnish methyl 3 -amino- 1H- pyrazole-5-carboxylate (14B)(4.4 g, 100%) as an off-white solid; mp 134.1 C; 1H NMR (300 MHz, DMSO) delta 12.99-1 1.69 (bs, IH), 5.77 (s, IH), 5.03 (bs, 2H), 3.74 (s, 3H). MS (ES+) 142.2(M+l). |
| 87% | With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 4h; | To a stirred solution of methyl 3-nitro-lH-pyrazole-5-carboxylate(A59, 25.0 g, 146.19 mmol) in methanol(500 mL) was added Pd/C(3.80 g, 36.54 mmol) under N2 atmosphere and the reaction mixture was stirred under H2 balloon pressure at room temperature for 4 h. After completion of reaction mixture filtered through celite bed and washed with EtOAc. The filtrate was concentrated to dryness. The residue was washed with n-pentane dried to get methyl 3-amino-lH-pyrazole-5-carboxylate A60 as a pale yellow solid. Yield: 18.0 g(87%) LC-MS(ES) m/z : 142.16[M+H]+. |
| With hydrogen;palladium 10% on activated carbon; In methanol; under 2585.81 Torr; | 123B. Preparation of methyl 3-amino-1H-pyrazole-5-carboxylate A suspension of <strong>[181585-93-3]methyl 3-nitro-1H-pyrazole-5-carboxylate</strong> (10 g, 58.4 mmol) and 10% Pd on charcoal (1.2 g) in methanol (150 Ml) was stirred under 50 psi pressure of hydrogen gas for 16 h. The reaction mixture was filtered over a bed of Celite and the filtrate was concentrated to give 9.2 g of methyl 3-amino-1H-pyrazole-5-carboxylate which was used as such in the next reaction. MS (ESI) m/z 142.3 (M+H). |
| With hydrogen;palladium 10% on activated carbon; In methanol; at 20℃; for 18h; | To 14.0 g <strong>[181585-93-3]methyl 3-nitro-1H-pyrazole-5-carboxylate</strong> in 200 ml methanol are added 1.2 g Pd/C (10% w/w). The mixture is stirred under H2-atmosphere at room temperature for 18 h. The mixture is filtrated over kieselgur. The filtrate is concentrated and the crude product is used without further purification.MS (M+1): 141Characteristic 1H NMR (300 MHz, dDMSO) signals: 5.7 ppm (s, 1H); 3.8 ppm (s, 3H) | |
| With hydrogen;palladium 10% on activated carbon; In methanol; at 20℃; for 18h; | Step3: Methyl 5,7-dihydroxy-6-phenylpyrazolo[1 ,5-a]pyrimidine-2-carboxylate; A solution of 5.Og Methyl 3-amino-1 H-pyrazole-5-carboxylate, 8.3ml_ diethyl- phenylmalonate and 5OmL diisopropylethylamine in 5OmL DMF was heated to 1500C for 40h. The solvent was removed, the solid residue was dissolved in 2- propanol the mixture was stirred for 3 hours. The desired product was filtered, dried and was used without further purification. MS (M+1): 286 Characteristic 1 H NMR (300MHz, d6-DMSO) signals: 6.0 (s, 1 H); 3.8 (s, 3H) | |
| 100 mg | With palladium 10% on activated carbon; ammonium formate; In ethanol; at 70℃; for 1h; | To a solution of methyl 3-nitro- 1H-pyrazole-5-carboxylate (500 mg) in Ethanol (0.25M) was added 10% Palladium on Carbon (0.1 eq) and ammonium fomiate (8 eq) and the reaction was heated for 1 hour at 70C then cooled to room temperature, filtered through celite, rinsed withMethanol and concentrated to dryness. The crude intermediate was suspended in DCM and extracted with water. To the organic layer was added 3 eq of MP-TsOH catch and release resin whereupon the mixture was stirred for 1 hour, filtered to collect resin then eluted with 7N Ammonia in MeOH and concentrated to dryness to afford 100 mg of methyl 3-amino-1H-pyrazole-5-carboxylate. To a solution of methyl3-amino-1H-pyrazole-5-carboxylate in dioxane was added di-tert-butyl dicarbonate (1.5 eq) and the reaction was heated at 120C overnight, then stirred at room temperature for 6 hours before the addition of imidazole (4.5 eq). The reaction mixture was subsequently refluxed at 130C for 2 hrs then stirred at room temperature overnight, concentrated to dryness, suspended in Ethyl acetate and extracted 3x with 0.25N HC1 solution, dried, filtered and concentrated to afford 427 mg ofmethyl 3-Qert-butoxycarbonylamino)-1H-pyrazole-5-carboxylate as a light pink solid. Similar to as described in General Procedure A, 3-(tert-butoxycarbonylamino)- 1H-pyrazole-5-carboxylate (200 mg) was reacted with 2-fluoro-4-iodo-pyridine to give methyl 5-(tert-butoxycarbonylamino)- 1 -(4-iodo-2-pyridyl)pyrazole-3-carboxylate. The crude material was used directly in subsequent reactions. To a solution of5-(tertbutoxycarbonylamino)-1-(4-iodo-2-pyridyl)pyrazole-3-carboxylate in DCM was added 4NHC1 in dioxane (10 eq). The reaction was stirred at room temperature for 30 minutes thenconcentrated to dryness to afford crude methyl5-amino-1-(4-iodo-2-pyridyl)pyrazole-3-carboxylate as the HC1 salt. Similar to as described inGeneral Procedure J, methyl 5-amino-1-(4-iodo-2-pyridyl)pyrazole-3-carboxylate was reacted toafford 90 mg 5-amino-1-(4-iodo-2-pyridyl)pyrazole-3-carboxylic acid which was used in the nextstep without purification. Similar as to described in General Procedure B, 5-amino-1-(4-iodo-2-pyridyl)pyrazole-3-carboxylic acid was reacted with ammonium chloride to afford 90 mg of 5-amino-1-(4-iodo-2-pyridyl)pyrazole-3-carboxamide which was used in the next reaction without purification.Similar to as described in General Procedure E, 5-amino-1-(4-iodo-2-pyridyl) pyrazole-3-carboxamide (90 mg) was reacted with (3R)-3-ethynyl-3-hydroxy-1-methyl-pyrrolidin-2-one to give 14mg of the title compound (15%). M-i-H = 341.0; 1H NMR (400 MHz, DMSO-d6) oe 8.44 (dd, J= 5.2, 0.8 Hz, 1H), 8.03- 8.00 (m,1H), 7.68 (s, 1H), 7.29, (dd, J= 5.1, 1.5 Hz, 1H), 7.21 (s, 1H), 6.89 (s, 2H), 6.63 (s, 1H), 5.72 (s,1H), 3.41 - 3.34 (m, 2H), 2.81 (s, 3H), 2.48 - 2.43 (m, 1H), 2.26 - 2.17 (m, 1H). |
| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 123A. Preparation of methyl 3-nitro-1H-pyrazole-5-carboxylate The compound was synthesized according to the procedure as reported (Synthesis, 12, 2003, p 1815). |
[ 5292-43-3 ]
[ 181585-93-3 ]
[ 886355-32-4 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| With caesium carbonate; In acetone; for 5h; | 2-tert-Butoxycarbonylmethyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong>: To a stirring mixture of delta-nitro^H-pyrazole-S-carboxylic acid methyl ester ( 3.5 gm, 20.5 mmol) dissolved in 150 ml of acetone was added cesium carbonate ( 8.01 gm, 24.6 mmol) followed by the addition of tert-butyl bromoacetate ( 3.61 ml). After 5 hours the solids were filtered and the mixture chromatographed on silica gel using 10% ethyl acetate/hexanes as the eluent to obtain 4.16 gm of pure title product. ESI M+1= 286. |
[ 75-03-6 ]
[ 181585-93-3 ]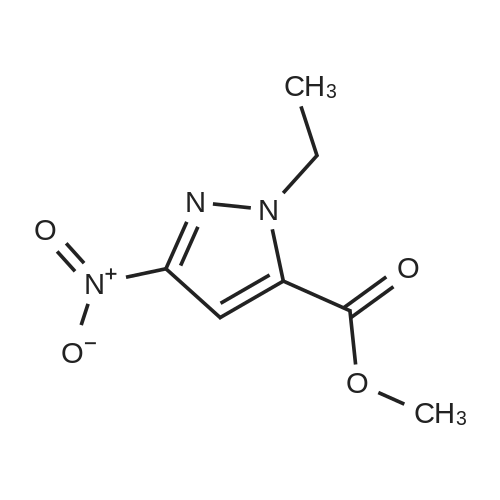 [ 1029052-28-5 ]
[ 1029052-28-5 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 44.7% | With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; | Step 2 - Preparation of2-ethyl-5-nitro-2H-pyrazote-3-carboxylic acid methyl ester (526):; [0137] To <strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> (525, 2.50 g, 0.0146 mol) in N,N- dimethylformamide (62.5 mL) were added iodoethane (1.2 mL, 0.016 mol) and potassium carbonate (4.17 g, 0.0301 mol) under an atmosphere of nitrogen. The reaction was stirred at room temperature overnight. The reaction was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography eluting with 20% to 100% ethyl acetate in hexane to give a white solid (526, 1.3 g, 44.7%). |
[ 67-56-1 ]
[ 181585-93-3 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 95% | With thionyl chloride; at 0℃;Reflux;Product distribution / selectivity; | Example 58 Acetic acid 2-[5-(5-azetidin-1-ylmethyl-1-methyl-1H-pyrazol-3-ylamino)-1-methyl-6-oxo-1,6-dihydro-pyridazin-3-yl]-6-(6-tert-butyl-8-fluoro-1-oxo-1H-phthalazin-2-yl)-benzyl ester To a solution of 5-nitro-1H-pyrazole-3-carboxylic acid (1.13 g, 7.19 mmol) in anhydrous methanol (20 ml) at to 0 C. was added thionyl chloride (2.23 g, 1.37 ml, 18.7 mmol) dropwise. The resulting solution was heated to reflux for 2 h. The cooled solution was evaporated to dryness to give 5-nitro-2H-pyrazole-3-carboxylic acid methyl ester (1.17 g, 95%) as a white solid. |
| With thionyl chloride; at 20℃; for 4h;Heating / reflux; | 9.Og 5-nitro-3-pyrazolo carboxylic acid are dissolved in abs. methanol and 7.6ml thionylchlohde are added dropwise at -100C. The reaction mixture is stirred at room temperature and refluxed for 4h. The solvent is evaporated and the crude product is used without further purification for the next step. MS (IVM ): 171 Characteristic 1 H NMR (300MHz, dDMSO) signals: 7.5ppm (s, 1 H); 3.9ppm (s, 3H) | |
| thionyl chloride; at -10 - 20℃; for 4h;Heating / reflux; | 9.0 g 5-nitro-3-pyrazolo carboxylic acid are dissolved in abs. methanol and 7.6 ml thionylchloride are added dropwise at -10 C. The reaction mixture is stirred at room temperature and refluxed for 4 h. The solvent is evaporated and the crude product is used without further purification for the next step.MS (M+1): 171Characteristic 1H NMR (300 MHz, dDMSO) signals: 7.5 ppm (s, 1H); 3.9 ppm (s, 3H) |
| With thionyl chloride; at -10 - 20℃;Reflux; | Intermediate Example 42.0 and 42.1 : Methyl 5-(4-formylphenyl)-6- phenylpyrazolo[1 ,5-a]pyrimidine-2-carboxylate and 5-(4-formylphenyl)-6- phenyl-pyrazolo[1 ,5-a]pyrimidine-2-carboxylic acid; Stepi : Methyl 3-nitro-1 H-pyrazole-5-carboxylate; 9.Og 5-nitro-3-pyrazolo carboxylic acid were dissolved in abs. methanol and 7.6ml_ thionylchloride were added dropwise at -100C. The reaction mixture was stirred at room temperature and refluxed for 4h. The solvent was evaporated and the crude product was used without further purification for the next step. MS (M+1): 171Characteristic 1 H NMR (300MHz1 d6-DMSO) signals: 7.5ppm (s, 1 H); 3.9ppm (s,3H) |
[ 181585-93-3 ]
[ 1225445-39-5 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 63% | With caesium carbonate; In acetonitrile; at 60℃; for 2h; | a) 5-Nitro-2-(3,4,5-trifluoro-benzyl)-2H-pyrazole-3-carboxylic acid methyl ester A mixture of methyl-3-nitro-1-H-pyrazole-5-carboxylate (ART-CHEM) (600 mg, 3.5 mmol), 3,4,5-trifluoromethylbenzyl bromide (789 mg, 3.5 mmol) and cesium carbonate (1.37 g, 4.2 mmol) in 15 ml of acetonitrile was stirred at 60 C. for 2 hours. The reaction mixture was concentrated, hydrolyzed and extracted with ethyl acetate. The organic phase was dried, evaporated and the residue triturated with diethyl ether to yield the title compound as a colorless solid (698 mg, Yield=63%). MS ISP (m/e): 333.1 (100) [(M+NH4)+]. 1H NMR (CDCl3, 300 MHz): delta (ppm)=7.43 (s, 1H), 7.04 (t, 2H), 5.75 (s, 2H), 3.95 (s, 3H). |
[ 181585-93-3 ]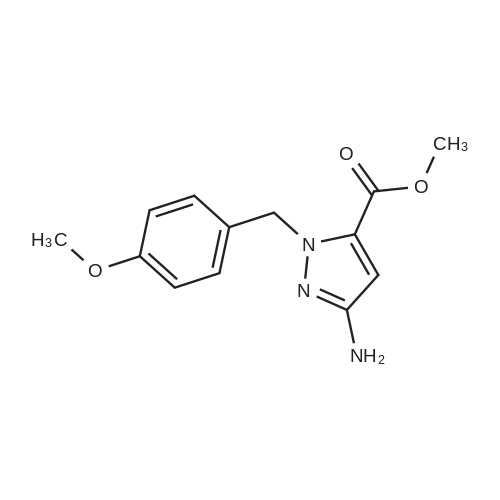 [ 625386-12-1 ]
[ 181585-93-3 ]
[ 1282606-65-8 ]
[ 181585-93-3 ]
[ 1282606-64-7 ]
[ 181585-93-3 ]
[ 1282606-67-0 ]
[ 181585-93-3 ]
[ 1282606-66-9 ]
[ 181585-93-3 ]
[ 1282606-68-1 ]
[ 181585-93-3 ]
[ 1282606-70-5 ]
[ 181585-93-3 ]
[ 625386-12-1 ]
[ 181585-93-3 ]
[ 1282606-65-8 ]
[ 181585-93-3 ]
[ 1282606-64-7 ]
[ 181585-93-3 ]
[ 1282606-67-0 ]
[ 181585-93-3 ]
[ 1282606-66-9 ]
[ 181585-93-3 ]
[ 1282606-68-1 ]
[ 181585-93-3 ]
[ 1282606-70-5 ]
[ 181585-93-3 ]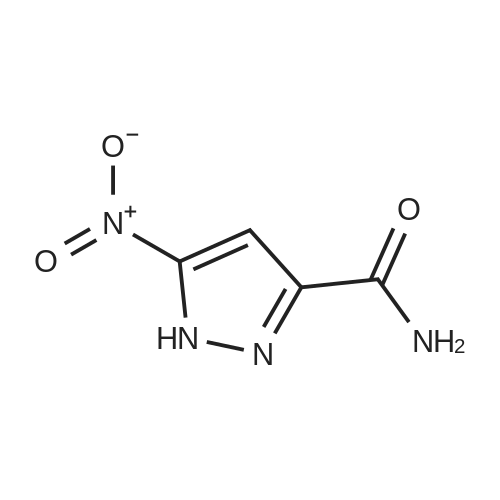 [ 297149-32-7 ]
[ 181585-93-3 ]
[ 1312929-43-3 ]
[ 181585-93-3 ]
[ 74-88-4 ]
[ 297149-32-7 ]
[ 181585-93-3 ]
[ 1312929-43-3 ]
[ 181585-93-3 ]
[ 74-88-4 ]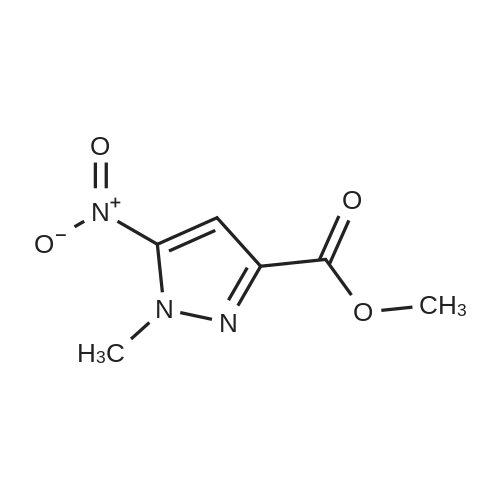 [ 796038-07-8 ]
[ 796038-07-8 ]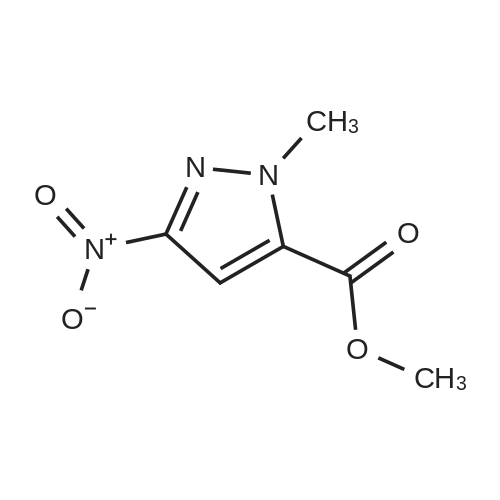 [ 177409-38-0 ]
[ 177409-38-0 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 18h; | To a solution of <strong>[181585-93-3]methyl 5-nitro-1H-pyrazole-3-carboxylate</strong> (1.87 g, 10.9 mmol) in anhydrous dimethyl formamide (20 mL) was added potassium carbonate (3.02 g, 21.9 mmol) and methyl iodide (2.02 g, 0.89 mL, 14.2 mmol) and the resulting solution stirred at room temperature for 18 h. The resulting mixture was diluted with water (1*150 mL) and extracted with dichloromethane (3*75 mL). The combined organic layers were dried over magnesium sulfate. The mixture was filtered and evaporated and the residue purified by flash chromatography (silica gel, 25 g, 20% to 60% dichloromethane in hexanes) to give a mixture of 2-methyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> and 1-methyl-5-nitro-1H-pyrazole-3-carboxylic acid methyl ester (1.64 g, 81%) as a white solid. | |
| With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 18h; | To a solution of <strong>[181585-93-3]methyl 5-nitro-1H-pyrazole-3-carboxylate</strong> (1.87 g, 10.9 mmol) in anhydrous dimethyl formamide (20 mL) was added potassium carbonate (3.02 g, 21.9 mmol) and methyl iodide (2.02 g, 0.89 mL, 14.2 mmol) and the resulting solution stirred at room temperature for 18 h. The resulting mixture was diluted with water (1 x 150 mL) and extracted with dichloromethane (3 x 75 mL). The combined organic layers were dried over magnesium sulfate. The mixture was filtered and evaporated and the residue purified by flash chromatography (silica gel, 25g, 20% to 60% dichloromethane in hexanes) to give a mixture of 2-methyl-5-nitro-2H-pyrazo le-3 -carboxylic acid methyl ester and 1-methyl-5-nitro-1H-pyrazole-3-carboxylic acid methyl ester (1.64 g, 81 %) as a white solid. | |
| With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 18h; | Step 12: Preparati -Methyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> A 100-mL single-neck round-bottomed flask was charged with methyl 5-nitro-lH-pyrazole-3- carboxylate (3.89 g, 22.7 mmol), anhydrous DMF (30 ml) , potassium carbonate (6.28 g, 45.5 mmol). Mel (4.19 g, 1.85 ml, 29.6 mmol) was added and the reaction mixture was stirred at room temperature for 18h. The mixture was then diluted with water (150 mL) and extracted with DCM (3 x 75mL). The combined organic layers were dried over MgS04 and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, AnaLogix system, SF40-240g column, 10% to 50% EtOAc in hexanes) to give the desired product as mixture of isomers (3.75 g). M+ = 185.0 m/e |
[ 181585-93-3 ]
[ 74-88-4 ]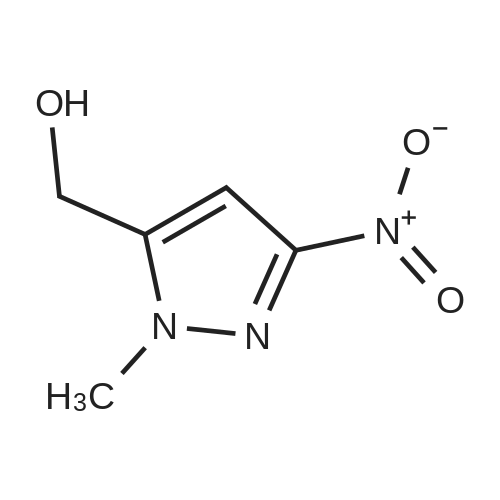 [ 1227210-46-9 ]
[ 1227210-46-9 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| To a solution of 2-methyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> and 1-methyl-5-nitro-1H-pyrazole-3-carboxylic acid methyl ester (1.18 g, 6.37 mmol) in tetrahydrofuran (20 mL) at 0 C. was added a lithium aluminum hydride solution (1.0M in tetrahydrofuran, 7.65 mL, 7.65 mmol) drop wise. The resulting mixture was stirred at 0 C. for 20 min. To this solution was added ethyl acetate (1 mL) followed by few crystals of sodium sulphate decahydrate. The resulting mixture was stirred for 30 min then filtered, the filter cake washed with ethyl acetate and the filtrate evaporated. The residue was purified by flash chromatography (silica gel, 80 g, 50% to 70% ethyl acetate in hexanes) to give 1-methyl-3-nitro-1H-pyrazol-5-yl)methanol (496 mg, 50%) as a white solid. | ||
| 496 mg | To a solution of 2-methyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> and 1-methyl-5-nitro-1H-pyrazole-3-carboxylic acid methyl ester (1.18 g, 6.37 mmo 1) in tetrahydro furan (20 mL) at 0C was added a lithium aluminum hydride solution (1 .OM in tetrahydrofuran, 7.65 mL, 7.65 mmol) drop wise. The resulting mixture was stirred at 0C for 20 min. To this solution was added ethyl acetate (1 mL) followed by few crystals of sodium sulphate decahydrate. The resulting mixture was stirred for 30 min then filtered, the filter cake washed with ethyl acetate and the filtrate evaporated. The residue was purified by flash chromatography (silica gel, 80 g, 50% to 70% ethyl acetate in hexanes) to give 1-methyl-3-nitro-1H-pyrazol-5-yl)methanol (496 mg, 50%) as a white solid. 1H NMR (300 MHz, DMSO-d6) delta ppm 3.90 (s, 3 H) 4.53 (d, J=5.67 Hz, 2 H) 5.55 (t, J=5.48 Hz, 1 H) 6.93 (s, 1 H). |
[ 106-93-4 ]
[ 181585-93-3 ]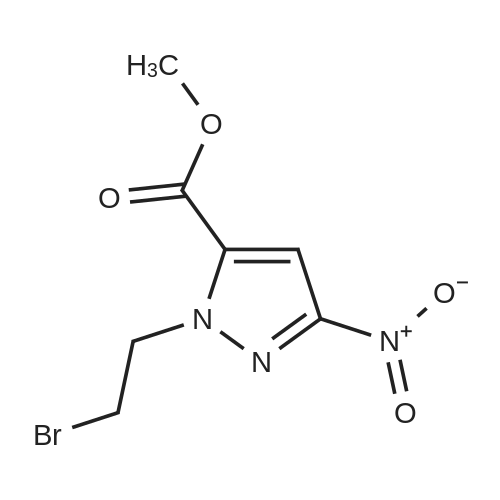 [ 1360057-00-6 ]
[ 1360057-00-6 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 100% | With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 100℃; for 4h; | To a stirred solution of 3-nitro-1H-pyrazole-5-carboxylate 20 (1.5 g, 0.87 mmol) in DMF (50 mL), cesium carbonate (5.7 g, 1.75 mmol) and then 1,2- dibromoethane (2.4 mL, 1.31 mmol) was charged portion wise at 0C. The reaction mixture was heated at 100C for 4 hour. The reaction mixture was diluted with dichioromethane and water (2x 100 mL). The combined organic extract was washed with water (3 x 30 mL). The organic layer was dried (Na2SO4), filtered and evaporated to obtain methyl 1 -(2-bromoethyl)-3 -nitro- 1 H-pyrazole-5-carboxylate 21(2.0 g, quantitative yield) as brown sticky solid. MS: 276.97 (M+H). |
| 92% | A 3 L 3-ne&ed flask fitted with refbax omleiver and t1nmcoap1er was chaiged with rn?thyl 3- nib-lH-pyraznle-5<a1boKy1ate (755 , 441 numl) anl DMF (735 mL). Cesium caJonate (173 , 52 nmvl) was a3dedpoi*xwise aid t1 rnchonwas hated to about 98 C fc5mizi, thencooled b antierdtemrerathre forabout3Uiuir. The ieacfioiiwas cooled inanicebathtoaboatU Cbefc the addifloit of 1,2-thlnnethare (380 niL, 4412 nmnol). The ieacthni was sthmd, wanuii to antierd temrenthr?, fc thout 5 K T& r?aDtioli nththm wa queirhel with tIE aMiboit of an aqueoas sobafioit of pcdassi?nu phcripha±e iuoiicbasic (120 iii 1 L). Th ieu1thtg thrriii was ethacted with ECAc (3 x 3D] mL). The combined oigaric poitoit was ded over MgSO4, fflteied azt orcenttated urder reluced pIes sure to affcd tMcavbcjyI (12] ?2%): LCIMS (Table l,Metlod I)R= 2.lU min.;MS ?th: 278, 230(M÷Hy. | |
| 77% | With potassium carbonate; In acetone; for 2h;Reflux; | To a solution ofmethyl3-nitro-1H-pyrazole-5-carboxylate (342 mg, 2.0 mmol) macetone (40 mL) was added 1,2-dibromoethane (412 mg, 2.2 mmol), followed by K2C03 (828mg, 6. 0 mmol). The resulting mixture was stirred to reflux for 2 hrs. Then K2C03 was filteredoff The filtrate was concentrated in vacuo to give a residue, which was purified by silica gelcolumn (DC.lV1/MeOH = 50/1) to afford methyll-(2-bromoethyl)-3-nitro-lH-pyrazole-5-carboxylate (430 mg, yield: 77%) as yellow solid.[001034] 1H NMR (400 rviHz, CDCb): J = 7.42 (s, 1H), 5.08 (t, J = 6.4 Hz, 2H), 3.97 (s, 3H),3.78 (t, J = 6.4 Hz, 2H). |
| 63% | With potassium carbonate; In acetone; for 2h;Reflux;Product distribution / selectivity; | To a solution of <strong>[181585-93-3]methyl 5-nitro-1H-pyrazole-3-carboxylate</strong> (1 g, 5.84 mmol) in acetone (30 mL) was added potassium carbonate (4.04 g, 29.2 mmol) and 1,2-dibromoethane (3.29 g, 1.51 mL, 17.5 mmol) and the resulting solution heated to reflux for 2 h. The resulting mixture was cooled to 0 C. filtered and concentrated and the residue purified by flash chromatography (silica gel, 60 g, 20% to 40% ethyl acetate in hexanes) to give methyl 1-(2-bromoethyl)-3-nitro-1H-pyrazole-5-carboxylate (1.03 g, 63%) as a white solid. |
| 41.1 mmol | With potassium carbonate; In acetone; at 60℃; for 1h; | Step b. A solution of methyl 3-nitro-lH-pyrazole-5-carboxylate (65.27 mmol) and K2CO3 (326.4 mmol) in acetone (335 ml) was stirred at rt for 20 min. 1,2-Dibromoethane (195.81 mmol) was added to the reaction mixture at rt and then stirred at 60C for 1 h. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The resulting residue was purified by column chromatography (9-10% EtOAc in hexane) yielding methyl l-(2-bromoethyl)-3-nitro-lH-pyrazole-5-carboxylate (41.10 mmol) MS: ES+ 278.05 (M) 280 (M+2); 1H NMR (400 MHz, DMSO-d6) delta ppm 7.63 (s, 1 H), 5.01 - 5.05 (m, 2 H), 3.92 - 3.96 (m, 2 H), 3.90 (s, 3 H). |
[ 106-93-4 ]
[ 181585-93-3 ]
[ 1360057-00-6 ]
[ 1431159-64-6 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| With potassium carbonate; In acetone; for 2h;Reflux; | To a solution of <strong>[181585-93-3]methyl 5-nitro-1H-pyrazole-3-carboxylate</strong> (5.97 g, 35 mmol) in acetone (100 mL) was added potassium carbonate (24 g, 174 mmol) and 1,2-dibromoethane (19.7 g, 9.02 mL, 105 mmol) and the resulting solution heated to reflux for 2 h. The resulting mixture was allowed to warm to room temperature over night, filtered and concentrated and the residue purified by flash chromatography (silica gel, 400g, 20% to 70% ethyl acetate in hexanes) to give a 11:2 mixture of methyl 1-(2-bromoethyl)-3-nitro-1H-pyrazole-5-carboxylate and 2-(2-Bromo-ethyl)-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> (4.86 g, 50%) as a light yellow solid. Major isomer component 1H NMR (300 MHz, DMSO-d6) delta ppm 3.77 - 4.05 (m, 40 H) 5.02 (t, J6.04Hz, 16 H) 7.61 (s, 5.65 H) 7.70 (s, 1 H). |
[ 106-93-4 ]
[ 181585-93-3 ]
[ 1227210-30-1 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| To a suspension of lithium borohydride (755 mg, 34.7 mmol) in tetrahydrofuran (100 mL) at 0C was added methyl 1-(2-bromoethyl)-3-nitro-1H-pyrazole-5-carboxylate (4.82 g, 17.3 mmol) in 10 mL of THF slowly. The resulting mixture was allowed to warm to room temperature 2 h. To the resulting mixture was added ethyl acetate (20 ml) and water (20 ml). The biphasic mixture was separated and aqueous layer extracted with ethyl acetate (3 x 20 mL). The combined organic layers were dried over magnesium sulfate and the resulting mixture was filtered and concentrated in vacuo. to give a crude mixture of 2-(2-Bromo-ethyl)-5-nitro-2H-pyrazol-3-yl]- methanol and (1-(2-bromoethyl)-3-nitro-1H-pyrazol-5-yl)methanol (4.24, 97%) as a light yellow oil, which was used as is in the next reaction. |
[ 106-93-4 ]
[ 181585-93-3 ]
[ 1227210-31-2 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 67% | To as solution of (1-(2-bromoethyl)-3-nitro-1H-pyrazol-5-yl)methanol (4.24 g, 17 mmol) in chloroform (100 mL), cooled to 0C, was added phosphorus tribromide (4.59 g, 1.6 mL, 17 mmol) drop wise. The resulting solution was warmed to room temperature and stirred for 2 h. The resulting solution was cooled to 0C and diluted with dichloromethane (50 ml). The resulting solution was made basic (pH 8.5) with saturated aqueous sodium bicarbonate was (20 mL). The layers were separated, and the aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed with brine (30 mL), dried over magnesium sulfate. The resulting mixture was filtered and concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, SF40-240 g, 15% to 40% EtOAc in hexanes) to give 1-(2-bromoethyl)-5-(bromomethyl)-3-nitro-1H-pyrazole (3.58 g, 67%) as a white solid. 1H NMR (300 MHz, DMSO-d6) delta ppm 3.92 (t, J=6.23 Hz, 2 H) 4.69 (t, J=6.23 Hz, 2 H) 4.89 (s, 2 H) 7.19 (s, 1 H). |
[ 181585-93-3 ]
[ 74-88-4 ]
[ 1227210-46-9 ] [ 1260659-40-2 ]
[ 1260659-40-2 ]| Yield | Reaction Conditions | Operation in experiment |
|---|---|---|
| 65.7%; 249 mg | Step 12: Preparati -Methyl-<strong>[181585-93-3]5-nitro-2H-pyrazole-3-carboxylic acid methyl ester</strong> A 100-mL single-neck round-bottomed flask was charged with methyl 5-nitro-lH-pyrazole-3- carboxylate (3.89 g, 22.7 mmol), anhydrous DMF (30 ml) , potassium carbonate (6.28 g, 45.5 mmol). Mel (4.19 g, 1.85 ml, 29.6 mmol) was added and the reaction mixture was stirred at room temperature for 18h. The mixture was then diluted with water (150 mL) and extracted with DCM (3 x 75mL). The combined organic layers were dried over MgS04 and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, AnaLogix system, SF40-240g column, 10% to 50% EtOAc in hexanes) to give the desired product as mixture of isomers (3.75 g). M+ = 185.0 m/e Step 13: In a 250 mL three-necked flask equipped with a thermometer and nitrogen inlet, LiBH4 (882 mg, 40.5 mmol) was combined with THF (30mL) to give a white suspension and cooled to 0 C using an ice bath. To this mixture, methyl l-methyl-3-nitro-lH-pyrazole-5-carboxylate (3.75g, 20.3 mmol) dissolved in THF (lOmL) was slowly added keeping the internal temperature at 0 C. After the addition was complete, the cooling bath was removed and the reaction mixture was stirred at room temperature for lh. A few drops of MeOH were then added and the reaction mixture was stirred for 2h. The reaction was cooled to 0 C using an ice bath and EtOAc (20 mL) was added followed by slow addition of water (100 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 x lOOmL). The combined extracts were washed with brine (100 mL), dried over MgS04 and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, Analogix system SF40-240g, 30% to 60% EtOAc in hexanes) to give separately the title compound (2.09 g, 65.7 %) and it isomer (249 mg, 7.82 g). (M+H)+ = 157.9 m/e |